El material genético de un virus es como su manual de instrucciones: nos dice cómo está estructurado y cómo se comporta al ingresar en el organismo de las personas. Por eso, a medida que el SARS-CoV-2 se sigue propagando en el mundo, una de las tareas cruciales de la comunidad científica es realizar en forma permanente su mapeo genómico, un estudio que puede describirse como dibujar el árbol genealógico de las mutaciones del coronavirus. Solo así será posible identificar si muestra cambios en su virulencia o si aparecen linajes más agresivos que otros.
Dentro de los más de 900 mil genomas que se han secuenciado del virus de la covid-19 hay miles de variantes únicas. Eso no quiere decir que sean peligrosas. Solamente algunas han demostrado un comportamiento distinto. Se les llama variantes de preocupación y hasta la fecha son tres: la del Reino Unido (B.1.1.7), la de Sudáfrica (B.1.351) y la de Manaos (P.1).
Sin embargo, científicos peruanos han dado a conocer una nueva variante, bautizada como C.37, cuya mutación es similar a los linajes de mayor transmisión del virus hallados en Brasil, Reino Unido y Sudáfrica, según el reporte publicado en la plataforma digital Virological.org, donde virólogos de todo el mundo discuten sus resultados preliminares.
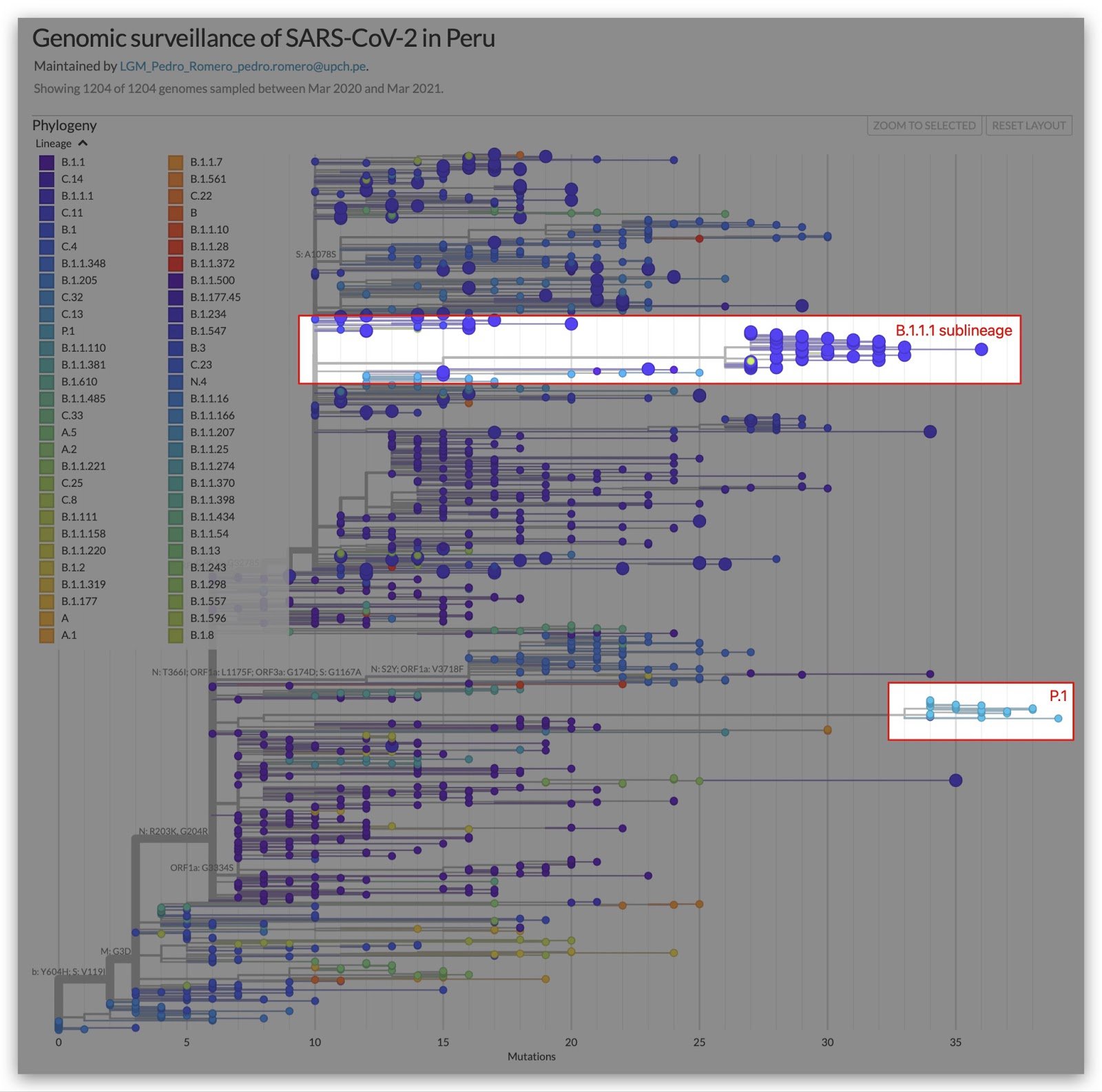
En la investigación - que deberá ser revisada por pares- participaron doce científicos de la Universidad Peruana Cayetano Heredia y de la Universidad Nacional Mayor de San Marcos. Este equipo señala que la nueva variante C.37, que desciende del linaje B.1.1.1 que circula por todo el mundo desde inicios de la pandemia, ha sido identificada en Perú y Chile, sin que esto quiera decir que se haya originado en uno de estos dos países, ya que es posible que provenga de otro lugar.
“Con los datos disponibles, no podemos asegurar si C.37 se originó en alguno de estos dos países. Ambos tienen epidemias similares en 2021 y comparten muchos vuelos diarios. Otra posibilidad es que se haya introducido a Chile y Perú desde otro país de la región que aún no la detecta”, detalló Pablo Tsukayama, investigador principal del estudio del mapeo genómico del SARS-CoV2.
La variante C.37 se ha propagado en Lima desde fines de diciembre de 2020 y parece expandirse rápidamente. Sus descubridores detallan que tiene una mutación conocida como deleción, que significa que ha perdido material genético. Pero, hasta ahora, la principal característica que genera preocupación es la mutación en la proteína de la espícula o «S» (del inglés spike, que significa punta y que le da forma de corona al SARS-CoV2), ya que podría volverla más contagiosa porque le ayudaría a evadir los efectos neutralizantes de los anticuerpos, una particularidad que también es compartida con la P.1.
Si bien la variante C.37 comparte mutaciones con los linajes del coronavirus P.1.(Brasil), B.1.1.7 (Reino Unido) y B.1351 (Sudáfrica), no es posible aún afirmar que es más contagiosa o más letal. Para saberlo serán necesarios más estudios, afirma el coautor del estudio, el biólogo Pedro Romero. “Se necesitan más datos, como, por ejemplo, cuántos pacientes (de los que poseen esta nueva variante) ingresaron a una unidad de cuidados intensivos. Eso toma más tiempo” añade.
La muestra realizada para el reporte preliminar implicó el análisis de 50 de 105 genomas del virus entre el 1 de enero y 18 de marzo en Lima.
Para Pablo Tsukayama es posible que muchas de las muestras identificadas por el Instituto Nacional de Salud (INS) como variante P.1 sean en realidad de la C.37. “El Ministerio de Salud no ha usado el secuenciamiento del genoma completo, sino una alternativa de menor costo, basada en una prueba PCR”, explica. Esto puede haber generado que se detecten ambas variantes sin diferenciarlas, ya que comparten la misma mutación. Por eso, el equipo investigador ya ha iniciado coordinaciones con el INS para evaluar esas muestras.
El último fin de semana, el ministro de Salud, Óscar Ugarte, recordó que el Instituto Nacional de Salud halló en marzo la presencia de la P.1 en 39.7% de las muestras tomadas en Lima. “Estamos atentos a la posibilidad de que puedan surgir otros linajes, hasta ahora no tenemos esa confirmación”, dijo. Sin embargo, cabe destacar que de los 105 genomas analizados para el reciente reporte, sólo el 2.8% eran de la P.1 frente al 43.1% de la C.37.
En el reporte preliminar, los investigadores señalan que, si bien durante estos meses se ha atribuido a la variante P.1 el aumento de contagios y muertes se abre ahora la posibilidad de que la segunda ola en Perú y Chile esté impulsada por la creciente prevalencia de la C.37. Ambos países, con procesos de vacunación en marcha, no han podido aún controlar el aumento significativo de contagios y hospitalizaciones. Sin embargo, es prematuro sacar conclusiones, pues se necesitan más estudios de corroboración.
En los últimos días, se ha conocido que la C.37 se ha propagado en Chile desde enero y hay reportes que confirman su presencia en Ecuador, Argentina, Brasil, Alemania, España, Estados Unidos y el Reino Unido. El último sábado, el Instituto de Microbiología de la Universidad San Francisco de Quito mostró preocupación por la presencia de cinco casos de la variante C.37 en la provincia costera de El Oro, fronteriza con el Perú, y en la andina de Pichincha, cuya capital es Quito.
¿Por qué es importante el hallazgo de nuevas variantes?
Aunque en los últimos años hemos oído hablar más sobre la secuenciación de genomas para la medicina personalizada, para la búsqueda de genes asociados a enfermedades neurodegenerativas o para detener la progresión de tumores, casi siempre nos referimos al genoma humano. Pero la realidad es que también secuenciamos, y en gran cantidad, los genomas de microorganismos patógenos.
En este caso, los genomas nos aportan otro tipo de información. El primer genoma de SARS-CoV-2 permitió identificar al virus como un coronavirus y comprobar que era diferente a los otros conocidos.
La mutación es un proceso natural. El virus acumula una o dos mutaciones por mes. Mientras más tiempo pase, el virus cambiará más. Pero también necesita espacio. Es decir, personas a quién contagiar. Tanto en Brasil como en Reino Unido y Sudáfrica hubo epidemias descontroladas. Por eso probablemente no escucharemos variantes peligrosas en Australia o Nueva Zelanda donde han aplastado al virus.
“Si no hay casos, no hay oportunidades de cambiar. Pero si hace poco tuvimos 11.260 nuevos contagios en un solo día es obvio que tenemos oportunidades de sobra para que el virus siga mutando”, explica Pablo Tsukayama. Esto es lo que ha pasado en los últimos meses en Perú y Chile.
Desde la primera ola, Tsukayama lidera un equipo que se dedica a estudiar los genomas del SARS-CoV-2 en el Perú, aunque con poco apoyo por parte del Estado. “Estamos haciendo muestras del 0.1% de los casos. Es insuficiente. Se nos escapan muchas variantes que están en Perú, porque el muestreo es muy bajo”, dijo hace unas semanas en una entrevista con Salud con lupa.
Con el mapeo genómico del virus se pueden tomar mejores decisiones sanitarias para controlar la pandemia. Por ejemplo, se pueden realizar mapas de cómo el virus se mueve de un lugar a otro; es posible estudiar si el virus de una persona que tiene un cuadro más severo tiene alguna característica particular; o si analizamos muchas muestras se puede detectar si el virus va mutando rápido e identificar a las variantes que se adaptan a una población. Además, el conocimiento de los linajes que circulan en un área geográfica permite evaluar si las actuales vacunas son efectivas o si será necesario desarrollar nuevas generaciones de vacunas para inmunizar a las personas.